在几十年的拖沓之后,FDA告知制药商,很快就会要求临床试验扩大入选范围,以便保证性别平衡、文化多样性和更宽泛的年龄范围等。看来已经无可避免,制药行业需要面对这一需求,主动设计临床试验,使之更加符合候选药物的目标人群。
主要难题
药企运营挑战
制药行业已经开始认真对待。生物科技行业组织(BIO)科学和法规事务主任Andrew Womack表示,在生物科技行业,更大的人口统计学纳入范围“肯定是优先考虑的事情”。因为提供数据证明基于性别、族裔或年龄具有遗传学差异的人对特定药物的应答情况,在科学上是正确的。不过,Womack承认,具体情况会因“治疗类型和公司”而异。
“制药商必须走得更远。”美国药品研究和制造商协会(PhRMA)科学和监管事务主任Jocelyn Ulrich也赞同招募更加多样化的患者人群。
但是,招募有代表性的亚群,有足够数量的患者人群才能得出正确结论,同时又要平衡更广泛人群的需求。对此,上海药明康德高级运营副总裁牟骅认为,在一个有特定选择的患者人群中开展临床研究,所得到的数据又该如何推广到更广泛的患者人群,这是一个常见问题。他承认,“那样会带来运营挑战。”
“我们正在面对年龄组的挑战,在过去,老年人群的代表性不足。”牟骅说。在加入药明康德之前,他担任过罗氏公司及基因泰克公司、Abraxis生物科学(现在的Celgene公司)、Biogen Idec公司的临床领导者。
招募失败
2014年6月,约翰斯霍普金斯大学医学院的研究者,在芝加哥举行的美国临床肿瘤学会会议上报告了一篇摘要,提示了“排除标准”和“运营障碍”对试验招募的影响。研究者根据在约翰斯霍普金斯开展的临床试验,分析了乳腺癌、非小细胞肺癌(NSCLC)、胰腺癌和前列腺癌患者的肿瘤学病历。
他们发现未能招募有4个最常见原因:没有可用试验或患者不符合合格性标准(36%~55%);建议或偏好标准治疗(6%~23%);患者住处离癌症中心太远无法参加(1%~22%);患者失访或选择在其他地方接受治疗(5%~14%)。合并后,这些因素共使64%的乳腺癌、83%的胰腺癌、84%的前列腺癌、89%的NSCLC患者不能参与在约翰斯霍普金斯开展的临床试验。
Womack建议,为了缓解其中一些问题,申办者应当考虑发起推广活动,为他们的研究塑造品牌,或许要联合当地医疗专业人员和社区组织,使人群增加熟悉程度。这些公司还应当仔细考虑如何解决一些公认的参与障碍,从初诊到儿童医疗到交通,并且考虑为这些服务提供补贴或付款。“有人可能认为这些步骤会增加费用,但如果周到考虑后,以战略性方式实施,也会带来巨大的价值。但付出成本,也会有机会。”
解决方案
“真实世界”目标
美国国立癌症研究所(NCI)综合癌症中心要求医疗组织开发网络,纳入来自社区服务区域的患者和供应者。癌症研究之友主席和创建者Ellen Sigal曾经帮助组织肺癌主方案(Lung-MAP),一项正在进行中的试验。
Lung-MAP试验有335个开放中心,目标是增加到500个中心,不仅要吸引大型学术性医疗中心,也要吸引遥远地区的小医院,地理分布要广泛。这项多药、生物标记物驱动的鳞细胞肺癌临床试验使用最先进的基因组分析技术,将患者与多项子研究相匹配。
为了研究相对罕见的癌症,必需有大型网络,才能招募充足的患者人群。Sigal强调,在患者居住的社区中找到他们,与改进癌症药物开发同等重要,因为大约85%的癌症治疗在社区进行,但这些患者经常未能获得临床试验选择。“我们需要获得这些在真实世界中接受治疗的患者。我相信,这些大型试验和NCI建立的这些新网络将达到目的。”
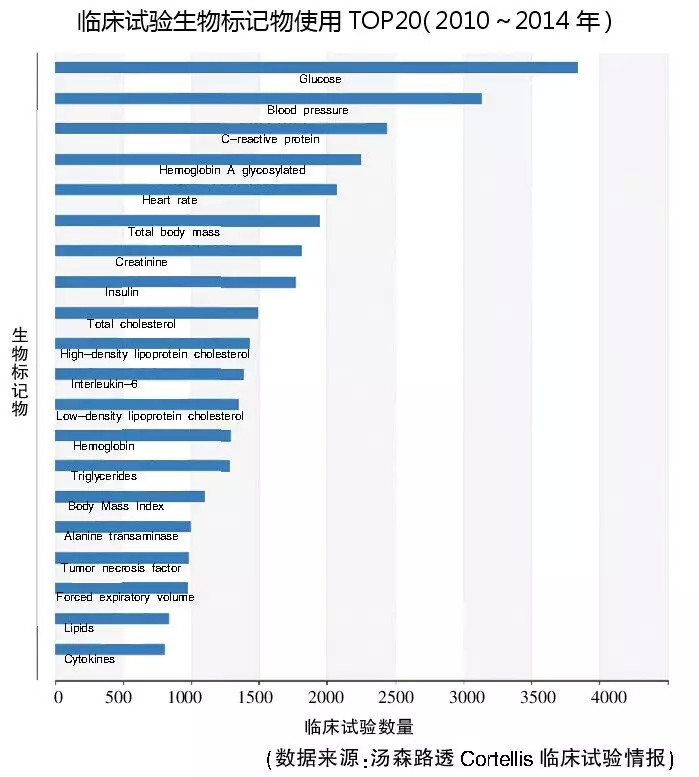
使用生物标记物
制药商可能会畏惧相应的成本增加,但昆泰公司临床开发总裁Paula Brown Staffor认为,费用不应该是试验人群更加多样化的阻碍因素。“我认为这不是一个费用问题,而只是在为合适的试验发现合适的患者。由于个体化药物的前景,招募族裔多样化的人群越来越受到重视。”
随着患者成为更加聪明的消费者,会寻求最佳疗法治疗疾病。患者向医生提出的问题超过从前,并且会评估医生对治疗方案中每种药物的了解程度如何,“药品处方信息要与患者组别相匹配,这越来越重要。”Stafford指出,生物标记物正在驱动这一过程。
$:page
根据Cortellis临床试验情报(CTI),自2000年以来,临床试验中的生物标记物使用增加了3倍以上,2000~2004年,开展了6620项;2005~2009年,开展了17,803项;2010~2014年,开展了28,821项。其中,肿瘤坏死因子和细胞因子等标记物的使用持续增长,属于前20名的生物标记物。
配备更好的检测工具,有可能大型药厂更加专业,而不是开发适合于所有患者的药物,这个趋势已经在开发罕见病用药的竞赛中变得很明显。
Stafford以适应性临床研究设计为例表示,其能使药物开发更加个体化,但不会增加临床试验的成本。“使用这样的方法开展临床试验,会比在有更广泛临床表现的更大患者人群中开展的临床试验小,也更集中。”包括使用有生物学意义和临床意义的替代指标作为非死亡率终点。“使用这种方法批准的药物说明书将清楚表明,该药的适应症较窄,只能用于明确限定的亚群,但已证明在这个亚群中药物的收益超过风险。”
她补充道,如果允许使用这样的设计和终点,应当要求申办者开展长期、许可后安全性和结局评价。
未必花更多钱
使用生物标记物、大数据和适应性设计等工具,意味着“不同方法完成相同任务”。应答率最高、疗效最高、安全性界限最大的人群,他们就是药物最安全最有效的患者。问题是:如何根据不同年龄、种族、性别使用生物标记物,发现相关的患者亚组?
而要了解不同地理区域、性别、疾病严重程度、人种、年龄之间的分布情况,需要严格监测患者分布,有时可启动额外的研究中心或关闭现有研究中心,以便达到合适的平衡。而这会花费更多的时间和更多的金钱。
对此,牟骅认为,寻求使用适应性试验设计与招募更广泛的人群并不是相互排斥的目标。实际上,使用适应性设计能够使申办者在亚群中对可能有前景的药物开展更有效率的试验。
他提供了假设的场景。“假设在一项试验中,有来自不同族裔组的患者,然后发现某个族裔组对药物的应答特别好。此时,你可能想修改研究设计,以这个研究亚群为目标人群,积累更多的数据,证明新药真正有益于这个人群。另一方面,如果药物对某个亚组根本没有益处,你可以快速做出决定,有可能剔除这个组。”
让医生小组多样化
Tufts药物开发研究中心(CSDD)资助项目主任和副教授、临床研究信息和研究中心主席Kenneth Getz指出,“文献显示,来自各种社区的少数民族患者参与临床试验的意愿非常高,但许多患者不知道也无法参加临床试验,因为很少有少数民族医生参与临床试验。”
Tufts CSDD研究发现,少数民族和女性主要研究者的比例非常低,而患者喜欢找与自己性别和族裔相同的医生就诊。“我们的研究显示,少数民族研究者流动率最高。”Getz建议,申办者和CRO可以投入资源与时间,去发现和培养与女性和少数民族医生、研究中心、网络的关系,以便包含更加多样化的医生小组。
“我们越了解药物如何在实际服用的患者中起效,整个行业的状况就会越好。从患者和公共卫生的角度,做正确的事情很重要。”Ulrich建议,制药商和CRO应当让社区医生了解试验运行机制,为他们提供充分的资源加入试验网络。此外,还应当与医疗领域之外但能接触服务匮乏人群的组织合作。
延伸<<<
FDA很认真!
一连串的指南、会议和备忘录在提示,FDA对改善临床试验人群的多样性很认真。
2005年,FDA发布了关于在临床试验中采集人种和族裔数据的行业指南在2012年的《FDA安全与创新法案》(FDASIA)第907节,扩大入选范围成为命令,要求在临床试验中纳入人口统计学亚组。
2013年12月,FDA药物评价和研究中心(CDER)在一份题为《审查管理规范:研究性新药申请的临床审查》的备忘录中,分析了临床试验中的老年患者入选情况。这项分析的结论是:申办者通常拒绝纳入患有多种慢性疾病的老年受试者,包括抑郁等精神障碍受试者、心脏病、动脉粥样硬化或糖尿病受试者。而其实,早在1989年,FDA就发布指南,要求在药物试验中入选更多的老年人。
2014年2月的一次电话会议中,CDER临床科学副主任Robert Temple建议,在讨论方案设计以及Ⅱ期试验结束会议期间,申办者必须准备好回答与广泛入选相关的问题。
2014年4月,FDA举行了一次公开会议,就改善新药申请中人口统计学亚组数据的采集、分析和可用性,收集行业反馈。来自各个组织的演讲者向FDA提供了大量建议,包括需要加强实施现有要求,使试验人群更有代表性,以及在这些试验中加入更多的亚组分析。
而美国卫生与公共服务部(HHS)和NIH近期的主要政策变化可能加速相关临床研究要求的实施。HHS发表了一份通知,要求研究者提交不能达到临床或安全性终点的研究性化合物试验结果。NIH则发表了一份政策草案,会将HHS数据报告要求应用到其资助的干预性试验。拟定的改变将要求几乎所有临床试验必须根据2007年FDA补充法案第Ⅷ条进行报告,要求研究者登记临床试验,包括早期阶段和扩展研究,以标准化格式向ClinicalTrials.gov提交总结结果,无论试验结局如何。